Research Topics Dr. Stefan Naumann
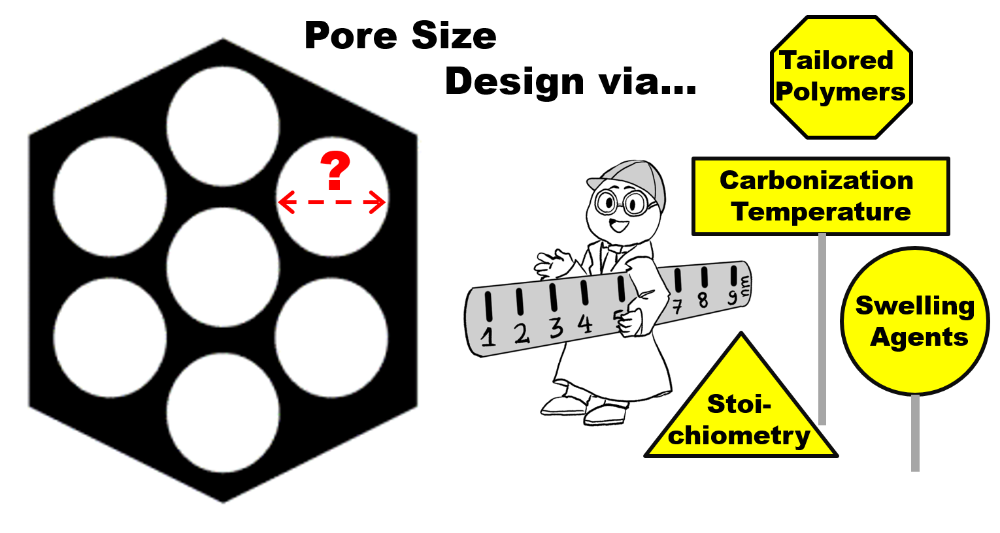
This research is conducted within the framework of CRC 1333 “Molecular Heterogeneous Catalysis in Defined Geometries”. The overarching target of this initiative is to exploit defined mesoporous support materials to host molecular catalysts (exclusively inside the pores) and impact their reactivity by exerting confinement effects and diffusion/transport phenomena. In this context, I am PI of a project investigating the porous supports, focusing on carbon materials. To this end, we employ an organic self-assembly process, using amphiphilic block copolyethers as structure directing agents. During a so-called EISA-process (Evaporation Induced Self-Assembly) different morphologies can be constructed (in our case 2D hexagonal), fixed (phenolic resin cross-linking) and carbonized (700°C, under nitrogen). Crucially, we employ our ability for precise polyether preparation (see 1-3) to in turn realize highly ordered carbon structures. By fine-tuning the structure-directing polymers with regard to molar mass and hydrophilic-to-lipophilic balance, we aim to control the diameter of the generated mesopores. We further aim to control the surface chemistry of the mesoporous material. In the second funding period of CRC 1333, self-supporting, ordered mesoprous carbon films/membranes are prepared, aiming for the electrocatalytic reduction of CO2.
See for example: https://www.crc1333.de/; A. Balint, M. Papendick, M. Clauss, C. Müller, F. Giesselmann, S. Naumann*, Chem. Commun. 2018, 54, 2220-2223; S. Naumann*, Org. Mater. 2021, 3, 283-294; F. Markus, C. Vogler, J. Bruckner, S. Naumann* ACS Appl. Nano Mater. 2021, 4, 3486-3492.
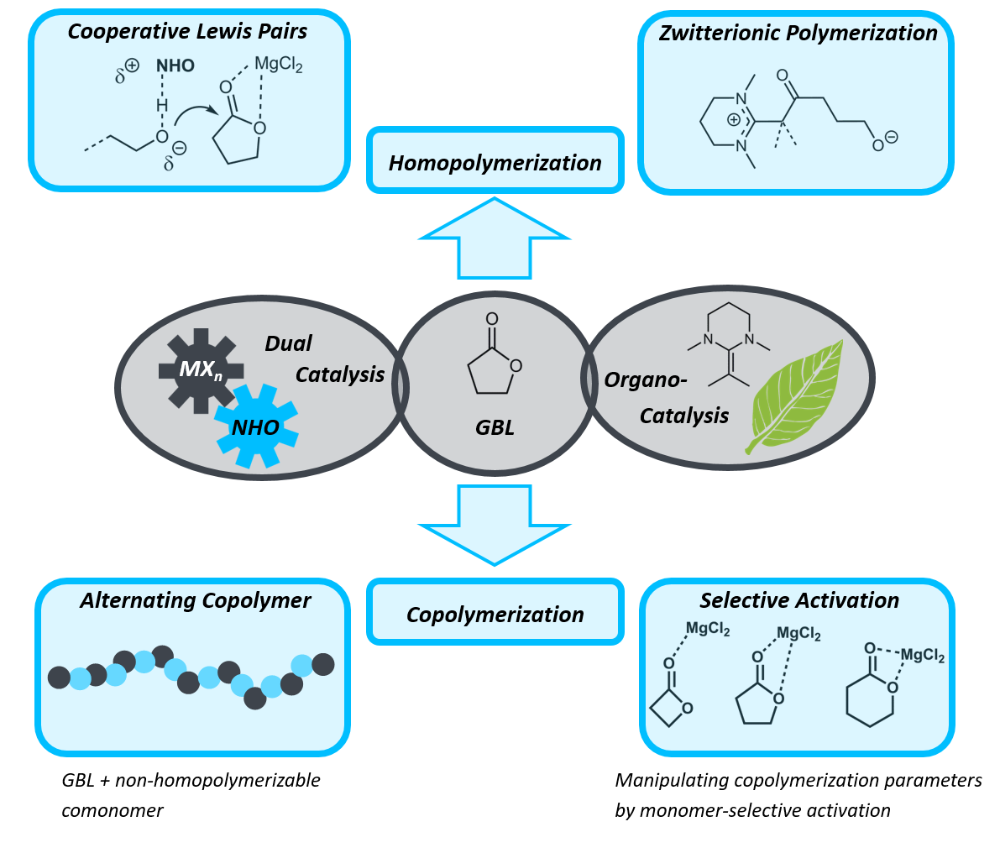
γ-Butyrolactone (GBL), as a rare exception among many monomers, combines several aspects which can render its application truly sustainable and economically attractive. This lactone can be bio-sourced and crucially, its thermodynamically favoured, (almost) strainless five-membered structure facilitates ready depolymerization (by simple heating). This typically occurs without any side products as demonstrated by Chen and co-workers, thus constituting an ideal candidate for a circular economy. However, to realize this aim, capable catalysis is required: until very recently, GBL was considered “non-polymerizable” and still remains challenging to do so. Our research therefore followed a two-pronged approach: developing more practicable ways to generate the homopolymer (poly(GBL)) and secondly copolymerizing GBL with other lactones to create tailored polyesters. In this case, the dual catalytic methodology proved especially successful, allowing for the selective realization of different poly(GBL) architectures and for controlling GBL incorporation in copolymers by choice of the co-catalyzing Lewis acid.
See for example: P. Walther, S. Naumann*, Macromolecules 2017, 50, 8406-8416; P. Walther, W. Frey, S. Naumann*, Polym. Chem. 2018, 9, 3674-3683.
A recent development in polymerization catalysis does not aspire to maximize the reactivity of, for example, the propagating chain end, but rather in opposite aims at employing propagating species with attenuated reactivity. This is counterbalanced by in turn activating the monomer. The benefits are especially notable if one achieves to selectively activate the monomer, while the polymer chain itself remains largely unaffected. To this end, my research activity has focused on developing simple, yet highly tunable catalyst pairings of Lewis bases (organobases) and Lewis acids for the ring-opening polymerization (ROP) of O-heterocyclic monomers. Multiple benefits have resulted from this approach, including among others:
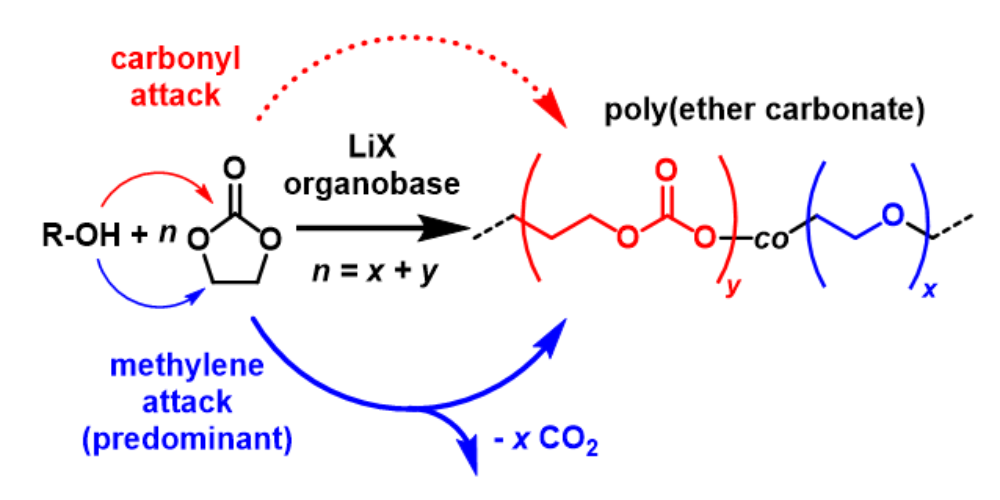
- A very rapid, yet highly selective (PDI < 1.0x) method for polymerizing lactones, where crucially the copolymerization parameters can be influenced, even switched by choice of the Lewis acid.
- The ability to chemically simplify both catalyst components, resulting in more competitive applications (i.e. ethylene carbonate polymerization, recycling applications).
- Realization of ultra-high molecular weight (UHMW) poly(propylene oxide) (Mn > 1 Mio. g/mol) and functional derivatives, by quantitatively suppressing transfer-to-monomer (see 1-3).
See for example: S. Naumann, P. B. V. Scholten, J. A. Wilson, A. P. Dove* J. Am. Chem. Soc. 2015, 137, 14439-14445; S. Naumann*, D. Wang Macromolecules 2016, 49, 8869-8878; P. Walther, A. Krauss, S. Naumann*, Angew. Chem. Int. Ed. 2019, 58, 10737-10741; N. von Seggern, T. Schindler, S. Naumann*, Biomacromolecules, 2020, 21, 2661-2669.
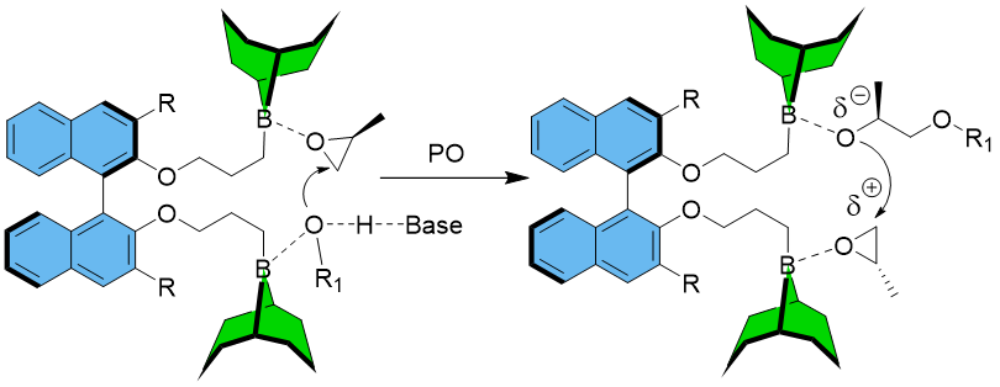
Two very different polymerization pathways, namely organocatalytic, anionic polymerization and zwitterionic polymerization can be operated by application of NHO catalysis. The different mechanisms have fundamental repercussions for the resulting polyethers. While in the former case highly defined polyether results (typically PDI ≤ 1.03), in the latter case ultra-high molar masses are generated (up to 1.6*106 g/mol), notably with the support of a Mg(II)-based monomer-activating agent. This mechanistic dualism opened up novel material applications for polyethers. As an example, we have used the metal-free route to synthesize amphiphilic block copolyethers (“Pluronics”-type) for creating improved hydrogels with a better mechanical robustness. Very differently, the resulting UHMW-polymers from the zwitterionic approach (i.e., poly(propylene oxide, PPO) are themselves very appealing (forming entangled elastomers, suitable for low-temperature application). The overall aim of my research regarding polyether formation is my conviction that, by perspective, polyethers can be developed into valuable performance polymers; to this end, a combination of advanced catalysis and process technology is necessary, encompassing not only control over molar mass and molar mass distribution, but also tacticity and degradability of the polyethers. In this regard, we have recently published the first example of a metal-free polymerization catalyst for the preparation of isotactoc-enriched PPO, using chiral diboranes.
See for example: S. Naumann*, A. W. Thomas, A. P. Dove* Angew. Chem. Int. Ed. 2015, 54, 9950-9954; P. Walther, A. Krauss, S. Naumann*, Angew. Chem. Int. Ed. 2019, 58, 10737-10741; F. Markus, J. Bruckner, S. Naumann*, Macromol. Chem. Phys. 2020, 221, 1900437; A. Sirin-Sariaslan, S. Naumann*, Chem. Sci. 2022, 13, 10939-10943.
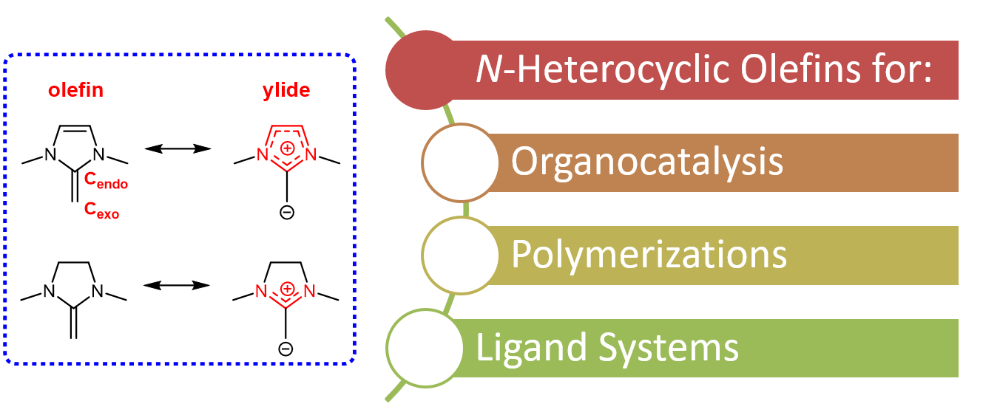
N-heterocyclic olefins (NHOs) constitute a class of very electron-rich, strongly polarized olefins. The heterocyclic backbone is prone to accommodating a positive (partial) charge, which results in charge separation; the corresponding electron excess is located on the exocyclic carbon atom. This renders the NHO a potent base and capable nucleophile, hence its general applicability in catalysis. During the past ~ 5 years, we have identified the crucial tuning parameters, enabling us to turn NHOs into powerful polymerization (co-)catalysts. Thus, the importance of aromatization was studied, rendering the backbone a crucial site for managing the NHO’s reactivity. Less obviously, but of comparable importance, is the impact of the N-substituents; beyond the expected stereoelectronic effects, these also control the planarity of the NHO and thus the degree of conjugation of its π electron system. By exploiting these properties, we succeeded in separately addressing nucleophilicity and basicity, thereby enabling different polymerization pathways. Overall, as we found from experiments and calculations, NHOs are superbases and can also add to moderately activated double bonds. Accordingly, we employ NHOs for the polymerization of epoxides, lactones, acrylic monomers and carbonates.
See for example: R. Schuldt, J. Kästner, S. Naumann* J. Org. Chem. 2019, 84, 2209-2218; S. Naumann* Chem. Commun. 2019, 55, 11658-11670.